
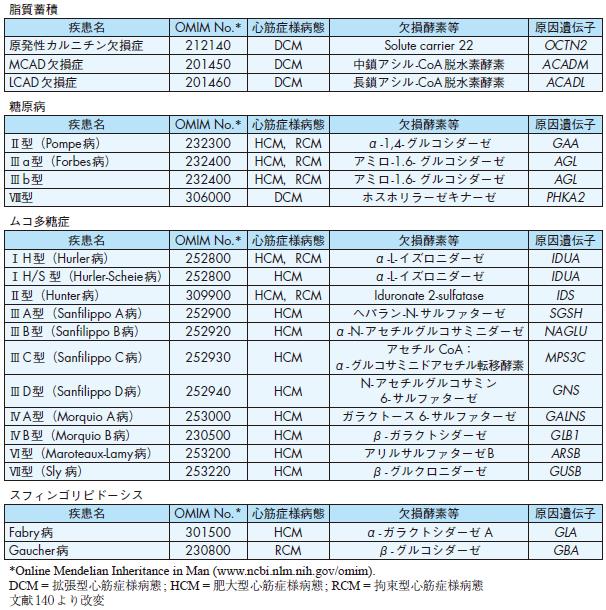
脂質蓄積
疾患名OMIM No.* 心筋症様病態欠損酵素等原因遺伝子
原発性カルニチン欠損症212140 DCM Solute carrier 22 OCTN2
MCAD欠損症201450 DCM 中鎖アシル-CoA脱水素酵素ACADM
LCAD欠損症201460 DCM 長鎖アシル-CoA脱水素酵素ACADL
糖原病
疾患名OMIM No.* 心筋症様病態欠損酵素等原因遺伝子
Ⅱ型(Pompe病) 232300 HCM,RCM α-1,4- グルコシダーゼGAA
Ⅲa型(Forbes 病) 232400 HCM,RCM アミロ-1.6- グルコシダーゼAGL
Ⅲb型232400 HCM,RCM アミロ-1.6- グルコシダーゼAGL
Ⅷ型306000 DCM ホスホリラーゼキナーゼPHKA2
ムコ多糖症
疾患名OMIM No.* 心筋症様病態欠損酵素等原因遺伝子
ⅠH型(Hurler病) 252800 HCM,RCM α-L- イズロニダーゼIDUA
ⅠH/S 型(Hurler-Scheie病) 252800 HCM α -L- イズロニダーゼIDUA
Ⅱ型(Hunter病) 309900 HCM,RCM Iduronate 2-sulfatase IDS
ⅢA型(Sanfilippo A病) 252900 HCM ヘパラン-N- サルファターゼSGSH
ⅢB型(Sanfilippo B病) 252920 HCM α-N- アセチルグルコサミニダーゼNAGLU
ⅢC型(Sanfilippo C病) 252930 HCM
アセチル CoA:α-グルコサミニドアセチル転移酵素MPS3C
ⅢD型(Sanfilippo D病) 252940 HCM N- アセチルグルコサミン
6- サルファターゼGNS
ⅣA型(Morquio A病) 253000 HCM ガラクトース 6- サルファターゼGALNS
ⅣB型(Morquio B病) 230500 HCM β- ガラクトシダーゼGLB1
Ⅵ型(Maroteaux-Lamy 病) 253200 HCM アリルサルファターゼB ARSB
Ⅶ型(Sly 病) 253220 HCM β- グルクロニダーゼGUSB
スフィンゴリピドーシス
疾患名OMIM No.* 心筋症様病態欠損酵素等原因遺伝子
Fabry病301500 HCM α- ガラクトシダーゼ A GLA
Gaucher病230800 RCM β- グルコシダーゼGBA
*Online Mendelian Inheritance in Man (www.ncbi.nlm.nih.gov/omim).
DCM = 拡張型心筋症様病態; HCM = 肥大型心筋症様病態; RCM = 拘束型心筋症様病態
文献140より改変
疾患名OMIM No.* 心筋症様病態欠損酵素等原因遺伝子
原発性カルニチン欠損症212140 DCM Solute carrier 22 OCTN2
MCAD欠損症201450 DCM 中鎖アシル-CoA脱水素酵素ACADM
LCAD欠損症201460 DCM 長鎖アシル-CoA脱水素酵素ACADL
糖原病
疾患名OMIM No.* 心筋症様病態欠損酵素等原因遺伝子
Ⅱ型(Pompe病) 232300 HCM,RCM α-1,4- グルコシダーゼGAA
Ⅲa型(Forbes 病) 232400 HCM,RCM アミロ-1.6- グルコシダーゼAGL
Ⅲb型232400 HCM,RCM アミロ-1.6- グルコシダーゼAGL
Ⅷ型306000 DCM ホスホリラーゼキナーゼPHKA2
ムコ多糖症
疾患名OMIM No.* 心筋症様病態欠損酵素等原因遺伝子
ⅠH型(Hurler病) 252800 HCM,RCM α-L- イズロニダーゼIDUA
ⅠH/S 型(Hurler-Scheie病) 252800 HCM α -L- イズロニダーゼIDUA
Ⅱ型(Hunter病) 309900 HCM,RCM Iduronate 2-sulfatase IDS
ⅢA型(Sanfilippo A病) 252900 HCM ヘパラン-N- サルファターゼSGSH
ⅢB型(Sanfilippo B病) 252920 HCM α-N- アセチルグルコサミニダーゼNAGLU
ⅢC型(Sanfilippo C病) 252930 HCM
アセチル CoA:α-グルコサミニドアセチル転移酵素MPS3C
ⅢD型(Sanfilippo D病) 252940 HCM N- アセチルグルコサミン
6- サルファターゼGNS
ⅣA型(Morquio A病) 253000 HCM ガラクトース 6- サルファターゼGALNS
ⅣB型(Morquio B病) 230500 HCM β- ガラクトシダーゼGLB1
Ⅵ型(Maroteaux-Lamy 病) 253200 HCM アリルサルファターゼB ARSB
Ⅶ型(Sly 病) 253220 HCM β- グルクロニダーゼGUSB
スフィンゴリピドーシス
疾患名OMIM No.* 心筋症様病態欠損酵素等原因遺伝子
Fabry病301500 HCM α- ガラクトシダーゼ A GLA
Gaucher病230800 RCM β- グルコシダーゼGBA
*Online Mendelian Inheritance in Man (www.ncbi.nlm.nih.gov/omim).
DCM = 拡張型心筋症様病態; HCM = 肥大型心筋症様病態; RCM = 拘束型心筋症様病態
文献140より改変
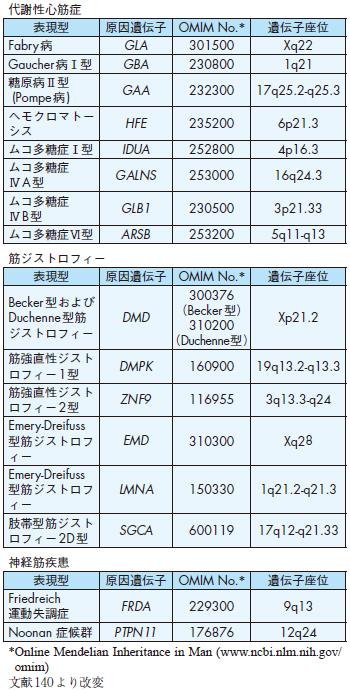
代謝性心筋症
表現型原因遺伝子OMIM No.* 遺伝子座位
Fabry病GLA 301500 Xq22
Gaucher 病Ⅰ型GBA 230800 1q21
糖原病Ⅱ型
(Pompe病) GAA 232300 17q25.2-q25.3
ヘモクロマトー
シスHFE 235200 6p21.3
ムコ多糖症Ⅰ型IDUA 252800 4p16.3
ムコ多糖症
ⅣA型GALNS 253000 16q24.3
ムコ多糖症
ⅣB型GLB1 230500 3p21.33
ムコ多糖症Ⅵ型ARSB 253200 5q11-q13
筋ジストロフィー
表現型原因遺伝子OMIM No.* 遺伝子座位
Becker 型および
Duchenne型筋ジストロフィー
DMD
300376(Becker 型)
310200(Duchenne型)
Xp21.2
筋強直性ジストロフィー1型DMPK 160900 19q13.2-q13.3
筋強直性ジストロフィー2型ZNF9 116955 3q13.3-q24
Emery-Dreifuss型筋ジストロフィー
EMD 310300 Xq28
Emery-Dreifuss型筋ジストロフィー
LMNA 150330 1q21.2-q21.3
肢帯型筋ジストロフィー2D型SGCA 600119 17q12-q21.33
神経筋疾患
表現型原因遺伝子OMIM No.* 遺伝子座位
Friedreich
運動失調症FRDA 229300 9q13
Noonan 症候群PTPN11 176876 12q24
* Online Mendelian Inheritance in Man (www.ncbi.nlm.nih.gov/omim)
文献140より改変
表現型原因遺伝子OMIM No.* 遺伝子座位
Fabry病GLA 301500 Xq22
Gaucher 病Ⅰ型GBA 230800 1q21
糖原病Ⅱ型
(Pompe病) GAA 232300 17q25.2-q25.3
ヘモクロマトー
シスHFE 235200 6p21.3
ムコ多糖症Ⅰ型IDUA 252800 4p16.3
ムコ多糖症
ⅣA型GALNS 253000 16q24.3
ムコ多糖症
ⅣB型GLB1 230500 3p21.33
ムコ多糖症Ⅵ型ARSB 253200 5q11-q13
筋ジストロフィー
表現型原因遺伝子OMIM No.* 遺伝子座位
Becker 型および
Duchenne型筋ジストロフィー
DMD
300376(Becker 型)
310200(Duchenne型)
Xp21.2
筋強直性ジストロフィー1型DMPK 160900 19q13.2-q13.3
筋強直性ジストロフィー2型ZNF9 116955 3q13.3-q24
Emery-Dreifuss型筋ジストロフィー
EMD 310300 Xq28
Emery-Dreifuss型筋ジストロフィー
LMNA 150330 1q21.2-q21.3
肢帯型筋ジストロフィー2D型SGCA 600119 17q12-q21.33
神経筋疾患
表現型原因遺伝子OMIM No.* 遺伝子座位
Friedreich
運動失調症FRDA 229300 9q13
Noonan 症候群PTPN11 176876 12q24
* Online Mendelian Inheritance in Man (www.ncbi.nlm.nih.gov/omim)
文献140より改変
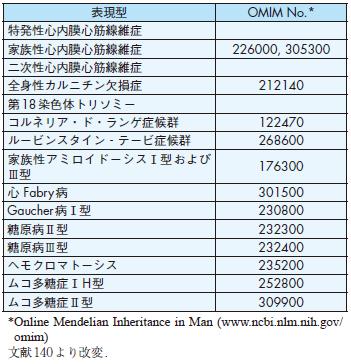
表現型OMIM No.*
特発性心内膜心筋線維症
家族性心内膜心筋線維症226000, 305300
二次性心内膜心筋線維症
全身性カルニチン欠損症212140
第18染色体トリソミー
コルネリア・ド・ランゲ症候群122470
ルービンスタイン‐テービ症候群268600
家族性アミロイドーシスⅠ型および
Ⅲ型176300
心 Fabry病301500
Gaucher病Ⅰ型230800
糖原病Ⅱ型232300
糖原病Ⅲ型232400
ヘモクロマトーシス235200
ムコ多糖症ⅠH型252800
ムコ多糖症Ⅱ型309900
* Online Mendelian Inheritance in Man (www.ncbi.nlm.nih.gov/omim)
文献140より改変.
特発性心内膜心筋線維症
家族性心内膜心筋線維症226000, 305300
二次性心内膜心筋線維症
全身性カルニチン欠損症212140
第18染色体トリソミー
コルネリア・ド・ランゲ症候群122470
ルービンスタイン‐テービ症候群268600
家族性アミロイドーシスⅠ型および
Ⅲ型176300
心 Fabry病301500
Gaucher病Ⅰ型230800
糖原病Ⅱ型232300
糖原病Ⅲ型232400
ヘモクロマトーシス235200
ムコ多糖症ⅠH型252800
ムコ多糖症Ⅱ型309900
* Online Mendelian Inheritance in Man (www.ncbi.nlm.nih.gov/omim)
文献140より改変.
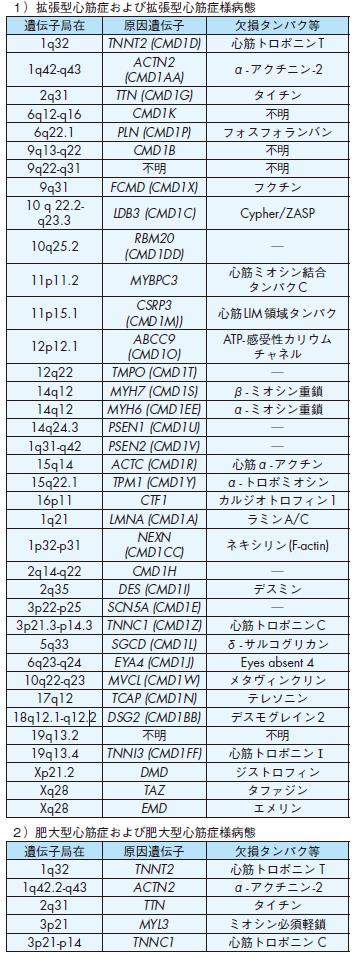
1)拡張型心筋症および拡張型心筋症様病態
遺伝子局在原因遺伝子欠損タンパク等
1q32 TNNT2 (CMD1D) 心筋トロポニンT
1q42-q43 ACTN2
(CMD1AA)
α- アクチニン-2
2q31 TTN (CMD1G) タイチン
6q12-q16 CMD1K 不明
6q22.1 PLN (CMD1P) フォスフォランバン
9q13-q22 CMD1B 不明
9q22-q31 不明不明
9q31 FCMD (CMD1X) フクチン
10 q 22.2-
q23.3 LDB3 (CMD1C) Cypher/ZASP
10q25.2 RBM20
(CMD1DD)
─
11p11.2 MYBPC3
心筋ミオシン結合
タンパクC
11p15.1 CSRP3
(CMD1M))
心筋LIM領域タンパク
12p12.1 ABCC9
(CMD1O)
ATP- 感受性カリウム
チャネル
12q22 TMPO (CMD1T) ─
14q12 MYH7 (CMD1S) β- ミオシン重鎖
14q12 MYH6 (CMD1EE) α- ミオシン重鎖
14q24.3 PSEN1 (CMD1U) ─
1q31-q42 PSEN2 (CMD1V) ─
15q14 ACTC (CMD1R) 心筋α- アクチン
15q22.1 TPM1 (CMD1Y) α- トロポミオシン
16p11 CTF1 カルジオトロフィン1
1q21 LMNA (CMD1A) ラミンA/C
1p32-p31 NEXN
(CMD1CC)
ネキシリン(F-actin)
2q14-q22 CMD1H ─
2q35 DES (CMD1I) デスミン
3p22-p25 SCN5A (CMD1E) ─
3p21.3-p14.3 TNNC1 (CMD1Z) 心筋トロポニンC
5q33 SGCD (CMD1L) δ- サルコグリカン
6q23-q24 EYA4 (CMD1J) Eyes absent 4
10q22-q23 MVCL (CMD1W) メタヴィンクリン
17q12 TCAP (CMD1N) テレソニン
18q12.1-q12.2 DSG2 (CMD1BB) デスモグレイン2
19q13.2 不明不明
19q13.4 TNNI3 (CMD1FF) 心筋トロポニンⅠ
Xp21.2 DMD ジストロフィン
Xq28 TAZ タファジン
Xq28 EMD エメリン
2)肥大型心筋症および肥大型心筋症様病態
遺伝子局在原因遺伝子欠損タンパク等
1q32 TNNT2 心筋トロポニン T
1q42.2-q43 ACTN2 α- アクチニン-2
2q31 TTN タイチン
3p21 MYL3 ミオシン必須軽鎖
3p21-p14 TNNC1 心筋トロポニン C
遺伝子局在原因遺伝子欠損タンパク等
1q32 TNNT2 (CMD1D) 心筋トロポニンT
1q42-q43 ACTN2
(CMD1AA)
α- アクチニン-2
2q31 TTN (CMD1G) タイチン
6q12-q16 CMD1K 不明
6q22.1 PLN (CMD1P) フォスフォランバン
9q13-q22 CMD1B 不明
9q22-q31 不明不明
9q31 FCMD (CMD1X) フクチン
10 q 22.2-
q23.3 LDB3 (CMD1C) Cypher/ZASP
10q25.2 RBM20
(CMD1DD)
─
11p11.2 MYBPC3
心筋ミオシン結合
タンパクC
11p15.1 CSRP3
(CMD1M))
心筋LIM領域タンパク
12p12.1 ABCC9
(CMD1O)
ATP- 感受性カリウム
チャネル
12q22 TMPO (CMD1T) ─
14q12 MYH7 (CMD1S) β- ミオシン重鎖
14q12 MYH6 (CMD1EE) α- ミオシン重鎖
14q24.3 PSEN1 (CMD1U) ─
1q31-q42 PSEN2 (CMD1V) ─
15q14 ACTC (CMD1R) 心筋α- アクチン
15q22.1 TPM1 (CMD1Y) α- トロポミオシン
16p11 CTF1 カルジオトロフィン1
1q21 LMNA (CMD1A) ラミンA/C
1p32-p31 NEXN
(CMD1CC)
ネキシリン(F-actin)
2q14-q22 CMD1H ─
2q35 DES (CMD1I) デスミン
3p22-p25 SCN5A (CMD1E) ─
3p21.3-p14.3 TNNC1 (CMD1Z) 心筋トロポニンC
5q33 SGCD (CMD1L) δ- サルコグリカン
6q23-q24 EYA4 (CMD1J) Eyes absent 4
10q22-q23 MVCL (CMD1W) メタヴィンクリン
17q12 TCAP (CMD1N) テレソニン
18q12.1-q12.2 DSG2 (CMD1BB) デスモグレイン2
19q13.2 不明不明
19q13.4 TNNI3 (CMD1FF) 心筋トロポニンⅠ
Xp21.2 DMD ジストロフィン
Xq28 TAZ タファジン
Xq28 EMD エメリン
2)肥大型心筋症および肥大型心筋症様病態
遺伝子局在原因遺伝子欠損タンパク等
1q32 TNNT2 心筋トロポニン T
1q42.2-q43 ACTN2 α- アクチニン-2
2q31 TTN タイチン
3p21 MYL3 ミオシン必須軽鎖
3p21-p14 TNNC1 心筋トロポニン C
7 心筋症
①原発性心筋症
1 )定義
従来,心筋症は“原因不明の心筋疾患”と定義されていたが,遺伝子解析等の進歩により,原因と推定されるものが多く発見された.それに伴い,1995年のWHO/
ISCF 委員会の定義は改訂され,心筋症は,“心機能障害を伴う心筋疾患”と定義され,“原因不明”の語句が外された102),103).心筋症の病因は多彩であり,機能と
形態に基づいた臨床病型による分類が用いられている.拡張型心筋症,肥大型心筋症,拘束型心筋症,不整脈原性右室心筋症の 4 型に分類され,これら4つの型に
入らない場合,分類不能型(unclassified)とされる.さらに,原因の明らかな疾患で,心筋症類似の病態を示す場合,特定心筋症と分類される.2006年のアメリカ心
臓協会(AHA)による心筋症の定義と分類では104),原因をより明確にする方向での分類が提唱されている.それによると,心筋症は原発性心筋症,二次性心筋症に
大別され,原発性心筋症はさらに,遺伝性心筋症,混合性(遺伝性および非遺伝性)心筋症,後天性心筋症に分類される.
Seidmanらによる肥大型心筋症における心筋βミオシン重鎖遺伝子の点突然変異の発見以来,心筋症での原因遺伝子の報告が世界各地で行われている105).しか
し,遺伝子変異を有しながら発症しない例や,臨床像との関連が少ない場合も多い.心筋症では遺伝子異常に加え,免疫異常やウイルス感染,さらに修飾遺伝子や
環境因子等の影響も指摘されており,複合的に発症することが予想されている.特定心筋症を除き,心筋症における遺伝子解析は研究段階にあるものも多く,現在の
ところ心筋症の診断は鑑別・除外診断が中心となる.しかしながら,将来的には原因遺伝子の同定がさらに進み,診断・治療に反映されることが期待される.
2 )拡張型心筋症
拡張型心筋症では,7 ~ 30 %に家族内発症が認められる105).常染色体優性遺伝の他,常染色体劣性遺伝,X染色体遺伝,ミトコンドリア異常が報告されている
106).拡張型心筋症の原因の一つとしてウイルス感染が指摘されており,家族内発症には感染症との関連も予想され,今後の課題である107),108).拡張型心筋症の
原因遺伝子として,dystrophin,lamin A/C等の他,βミオシン重鎖,トロポニンT等のサルコメア構成タンパクの遺伝子変異も報告されている(表4).
3 )肥大型心筋症
肥大型心筋症の約半数は常染色体優性遺伝の家族内発症であり,このような家族性肥大型心筋症は心筋サルコメア疾患と捉えられている.心筋βミオシン重鎖遺
伝子(MYH7)をはじめ,10以上の遺伝子の変異が報告されている(表4)105),109).タイチンの変異も報告されている110),111).
4 )拘束型心筋症
拘束型心筋症は広義には原因が不明な拘束型心筋症だけでなく,心アミロイドーシス,心内膜心筋線維症等が含まれる.原因としては遺伝子異常の場合とそれ以
外のものとがあるが,遺伝子異常によるものの大部分は代謝異常疾患である(表5).
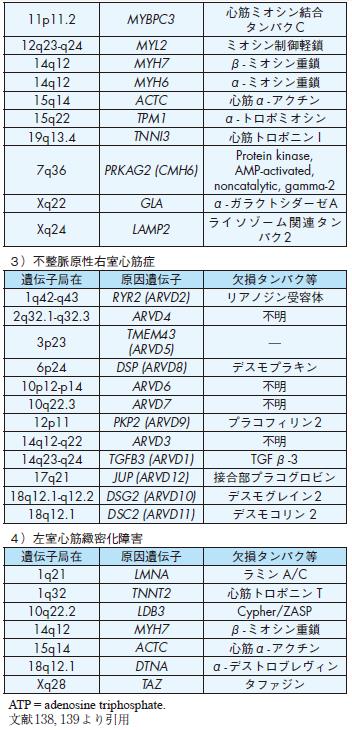
5 )不整脈原性右室心筋症
不整脈原性右室心筋症の約1/3は遺伝性と考えられ,常染色体優性または常染色体劣性遺伝形式をとる.原因遺伝子としてdesmoplakin 遺伝子,plakoglobin遺
伝子,リアノジン受容体遺伝子(RYR2),plakophilin-2遺伝子等が報告されている(表4)112)-121).
6 )左室心筋緻密化障害
左室心筋緻密化障害は,胎生期において左室心筋の粗な心内膜心筋が緻密な心筋構造になっていく過程が障害される先天性疾患の1つである.心内膜で覆われ
た肉柱が遺残し,心不全を発症し予後不良である.顔貌異常や他の心形成異常を伴わないものがある.近年,左室心筋緻密化障害を心筋症の1 つとする意見も増え
つつある.常染色体優性遺伝,X染色体遺伝が知られている.原因遺伝子として,X染色体上のtafazzin遺伝子が最初に報告されたが,現在では複数の遺伝子変異
が報告されている(表4)122).
②特定心筋症
1 )代謝性心筋症
①ミトコンドリア心筋症
ミトコンドリア心筋症はミトコンドリア病に含まれ,肥大型心筋症,拡張型心筋症,拘束性心筋症等の病態を呈する疾患群である123).ミトコンドリアの機能障害に
よるため,広義の代謝性心筋症に分類される.患者数は500~ 600例と推定されている.ミトコンドリア心筋症はミトコンドリアタンパクをコードする核DNAあるいは
mtDNA自体の変異に基づく疾患である.心筋症の病因となることが確認されている遺伝子変異,あるいは病因となることが確実と推定される遺伝子変異として,ミ
トコンドリア tRNA-Leu(UUR)遺伝子(3343A>G,3244G > A,3252A > G,3260A > G,3271T > C,3291T>C),ミトコンドリア tRNA-Val 遺伝子(1642G
>A),ミトコンドリア tRNA-Cys 遺伝子(5814A>G),ミトコンドリア tRNA-Lys 遺伝子,ミトコンドリアtRNA-Ile 遺伝子,ミトコンドリア COXⅢ 遺伝子(9957T
>C),ミトコンドリア ND5 遺伝子(13513G>A)等が報告されている124)-126).
遺伝性,家族性にあらわれるものがあるが,散発例も多い.ミトコンドリア DNA の既知の点変異を調べる検査には,血液試料または骨格筋等が用いられる.ミトコ
ンドリアゲノムの特性(ヘテロプラスミー)からミトコンドリアDNA変異の存在は必ずしも発症を意味しない.
一般に低年齢で発症した症例ほど予後不良で,成人例では緩徐に進行する.原因遺伝子変異と予後との関連は不明である.根本的治療法は確立していない.理
論的には変異 mtDNA を選択的に除去する方法が考えられるが,心筋では困難と思われる.カルニチン,ユビキノン等の治療法が行われてきたが,不十分であり,ま
た,原因遺伝子変異と治療効果との関連は不明である.海外ではミトコンドリア心筋症患者に対して心臓移植が行われているが,日本では行われていない.
ミトコンドリア心筋症の遺伝子解析は研究室レベルで行われており,一部の遺伝学的検査は東洋紡,BML,SRL, 苫小牧臨床検査センター等で行われている.
mtDNA は母系遺伝するため,患者本人に mtDNA の点変異の存在が判明した場合,母や母方親族に対する配慮が必要となる.
②心Fabry 病
Fabry病はα-galactosidase Aの遺伝子異常によるα-galactosidase A活性低下により生じ, 心Fabry病はFabry病の一病型(亜型)である(表 6,7).心Fabry
病では,心臓の細胞にスフィンゴ糖脂質が蓄積し,心肥大を来たすが,心臓以外の臓器障害やそれに伴う症状を欠く127).
α -galactosidase A遺伝子は X染色体のXq22 領域に存在するため,心Fabry病はX染色体劣性の遺伝形式をとる. 心Fabry病では,Ala20Pro,Glu66Gln,
Ile92The,Phe113Leu,Asn215Ser,Gln279Glu,Met296Ile,Met296Val,Arg301Gln,Tyr313Asp,Thr317Ile等のミスセンス変異が報告されている127)-130).発端
者の遺伝学的検査(2008年4 月より保険適用)にはα-galactosidase A遺伝子全長を調べる必要があり,変異が同定された場合は,その変異がα -galactosidase A
酵素活性低下を引き起こすことを確認する必要がある.
近年,本症に対する根本治療の1 つと考えられる遺伝子組み換えヒトα-galactosidase A酵素タンパクを用いた酵素補充療法が開発され,我が国においては2004年
4月から一般臨床応用が可能となった.心病変に関しては初期の病変に対する有用性が報告されている[レベルB].進行した心病変に対する有用性は十分に確認さ
れていない.早期診断・治療により,心病変の発症が予防され,また,心病変の進行を遅らせることができると予想されている.遺伝学的検査および酵素活性測定が
診断に重要である.予後や治療と遺伝子変異との関連については不明である.
③ Danon 病
Danon病はライソゾーム病の一種で, X染色体劣性遺伝.心肥大とミオパチーが主症状で精神発達遅滞を伴う.ライソゾーム膜の構成タンパク( Lysosome-
associated membrane protein 2)の異常でLAMP2変異による(表4)131).
④糖原病
グリコーゲン代謝経路の遺伝的異常に基づく疾患でグリコーゲン貯留性心肥大を来たす.糖原病Ⅱ型(Pompe病)(acid maltase欠損:17q21-q23),Ⅲ a型
(debrancher enzyme欠損:1p21),Ⅲb型(debrancher enzyme 欠損:1p21)等において心筋症様病態をとることが報告されている(表6,7)132).
⑤ムコ多糖症
いずれも常染色体劣性遺伝であり,弁膜症が主体であるが,左室収縮能低下や肥大型心筋症様病態を認める場合もある(表6,7).Hurler症候群(MPSⅠ:α - イ
ズロニダーゼ欠損症),Sanfilippo症候群(MPSⅢ:ヘパラン-N- サルファターゼ(A型),α -N- アセチルグルコサミニダーゼ(B型),アセチルCoA:α - グルコサミニ
ドアセチル転移酵素(C型),N- アセチルグルコサミン6- サルファターゼ(D型)の4 種の酵素欠損),Morquio症候群(MPSⅣ:ガラクトース6- サルファターゼ(A型),
β - ガラクトシダーゼ(B型) の2種の酵素欠損),Maroteaux-Lamy症候群(MPSⅥ:N- アセチルガラクトサミン4- サルファターゼ欠損),Sly病(MPSⅦ:β- グ
ルクロニダーゼ欠損症)で,心筋症様病態をとることが報告されている133).原因遺伝子が同定されており,遺伝学的検査が可能である.
⑥脂質蓄積
カルニチン欠損症等で報告されており,拡張型心筋症様病態をとる134)(表7).
⑦ヘモクロマトーシス
原発性(遺伝性)ヘモクロマトーシスの原因遺伝子は,6 番染色体短腕に位置するHFE遺伝子であり,拡張型心筋症様と拘束型心筋症様の混在した病態をとり得る
135)(表6).
2 )筋ジストロフィ―
Duchenne型および Becker型筋ジストロフィーはX染色体劣性遺伝形式をとり,ジストロフィン遺伝子異常による136)(表6).患者の遺伝子診断が2006年4月から健
康保険の適用となった(検査受託業者で検査可能).女性が保因者となるため配慮が必要である.筋強直性筋ジストロフィーは常染色体優性遺伝形式をとり,1型は
19番染色体長腕でのCTG リピートの異常伸長による疾患である.いずれも拡張型心筋症様病態を発症し得る.
3 )神経筋疾患
Friedreich運動失調症は常染色体劣性遺伝性で,欧米では最も多い脊髄小脳変性症であるが,日本では遺伝子レベルで診断が確定した例はまだない(表6).肥
大型心筋症様または拡張型心筋症様病態をとる場合がある.
Noonan 症候群の50%は12q24.1領域のプロテインチロシンリン酸化酵素遺伝子(PTPN11)の変異による.肥大型心筋症様病態をとる場合がある137).
表4 心筋症および心筋症様病態の原因遺伝子
11p11.2 MYBPC3
心筋ミオシン結合
タンパクC
12q23-q24 MYL2 ミオシン制御軽鎖
14q12 MYH7 β- ミオシン重鎖
14q12 MYH6 α- ミオシン重鎖
15q14 ACTC 心筋α- アクチン
15q22 TPM1 α- トロポミオシン
19q13.4 TNNI3 心筋トロポニン I
7q36 PRKAG2 (CMH6)
Protein kinase,
AMP-activated,
noncatalytic, gamma-2
Xq22 GLA α- ガラクトシダーゼA
Xq24 LAMP2
ライソゾーム関連タン
パク2
3)不整脈原性右室心筋症
遺伝子局在原因遺伝子欠損タンパク等
1q42-q43 RYR2 (ARVD2) リアノジン受容体
2q32.1-q32.3 ARVD4 不明
3p23 TMEM43
(ARVD5)
-
6p24 DSP (ARVD8) デスモプラキン
10p12-p14 ARVD6 不明
10q22.3 ARVD7 不明
12p11 PKP2 (ARVD9) プラコフィリン2
14q12-q22 ARVD3 不明
14q23-q24 TGFB3 (ARVD1) TGFβ-3
17q21 JUP (ARVD12) 接合部プラコグロビン
18q12.1-q12.2 DSG2 (ARVD10) デスモグレイン2
18q12.1 DSC2 (ARVD11) デスモコリン 2
4)左室心筋緻密化障害
遺伝子局在原因遺伝子欠損タンパク等
1q21 LMNA ラミン A/C
1q32 TNNT2 心筋トロポニン T
10q22.2 LDB3 Cypher/ZASP
14q12 MYH7 β- ミオシン重鎖
15q14 ACTC 心筋α- アクチン
18q12.1 DTNA α- デストロブレヴィン
Xq28 TAZ タファジン
ATP = adenosine triphosphate.
文献138, 139より引用
心筋ミオシン結合
タンパクC
12q23-q24 MYL2 ミオシン制御軽鎖
14q12 MYH7 β- ミオシン重鎖
14q12 MYH6 α- ミオシン重鎖
15q14 ACTC 心筋α- アクチン
15q22 TPM1 α- トロポミオシン
19q13.4 TNNI3 心筋トロポニン I
7q36 PRKAG2 (CMH6)
Protein kinase,
AMP-activated,
noncatalytic, gamma-2
Xq22 GLA α- ガラクトシダーゼA
Xq24 LAMP2
ライソゾーム関連タン
パク2
3)不整脈原性右室心筋症
遺伝子局在原因遺伝子欠損タンパク等
1q42-q43 RYR2 (ARVD2) リアノジン受容体
2q32.1-q32.3 ARVD4 不明
3p23 TMEM43
(ARVD5)
-
6p24 DSP (ARVD8) デスモプラキン
10p12-p14 ARVD6 不明
10q22.3 ARVD7 不明
12p11 PKP2 (ARVD9) プラコフィリン2
14q12-q22 ARVD3 不明
14q23-q24 TGFB3 (ARVD1) TGFβ-3
17q21 JUP (ARVD12) 接合部プラコグロビン
18q12.1-q12.2 DSG2 (ARVD10) デスモグレイン2
18q12.1 DSC2 (ARVD11) デスモコリン 2
4)左室心筋緻密化障害
遺伝子局在原因遺伝子欠損タンパク等
1q21 LMNA ラミン A/C
1q32 TNNT2 心筋トロポニン T
10q22.2 LDB3 Cypher/ZASP
14q12 MYH7 β- ミオシン重鎖
15q14 ACTC 心筋α- アクチン
18q12.1 DTNA α- デストロブレヴィン
Xq28 TAZ タファジン
ATP = adenosine triphosphate.
文献138, 139より引用
表5 拘束型心筋症関連疾患
表6 特定心筋症の原因遺伝子および遺伝子座位
表7 代謝性心筋症と原因遺伝子

心臓血管疾患における遺伝学的検査と遺伝カウンセリングに関する
ガイドライン(2011年改訂版)
Guidelines for Genetic Test and Genetic Councelling in Cardiovascular Disease(JCS 2011)
ガイドライン(2011年改訂版)
Guidelines for Genetic Test and Genetic Councelling in Cardiovascular Disease(JCS 2011)